Abstract
Background Genome-wide association studies (GWAS) have uncovered thousands of genetic variants that are associated with complex human traits and diseases. miRNAs are single-stranded non-coding RNAs. In particular, genetic variants located in the 3’UTR region of mRNAs may play an important role in gene regulation through their interaction with miRNAs. Existing studies have not been thoroughly conducted to elucidate 3’UTR variants discovered through GWAS. The goal of this study is to analyze patterns of GWAS functional variants located in 3’UTRs about their relevance in the network between hosting genes and targeting miRNAs, and elucidate the association between the genes harboring these variants and genetic traits.
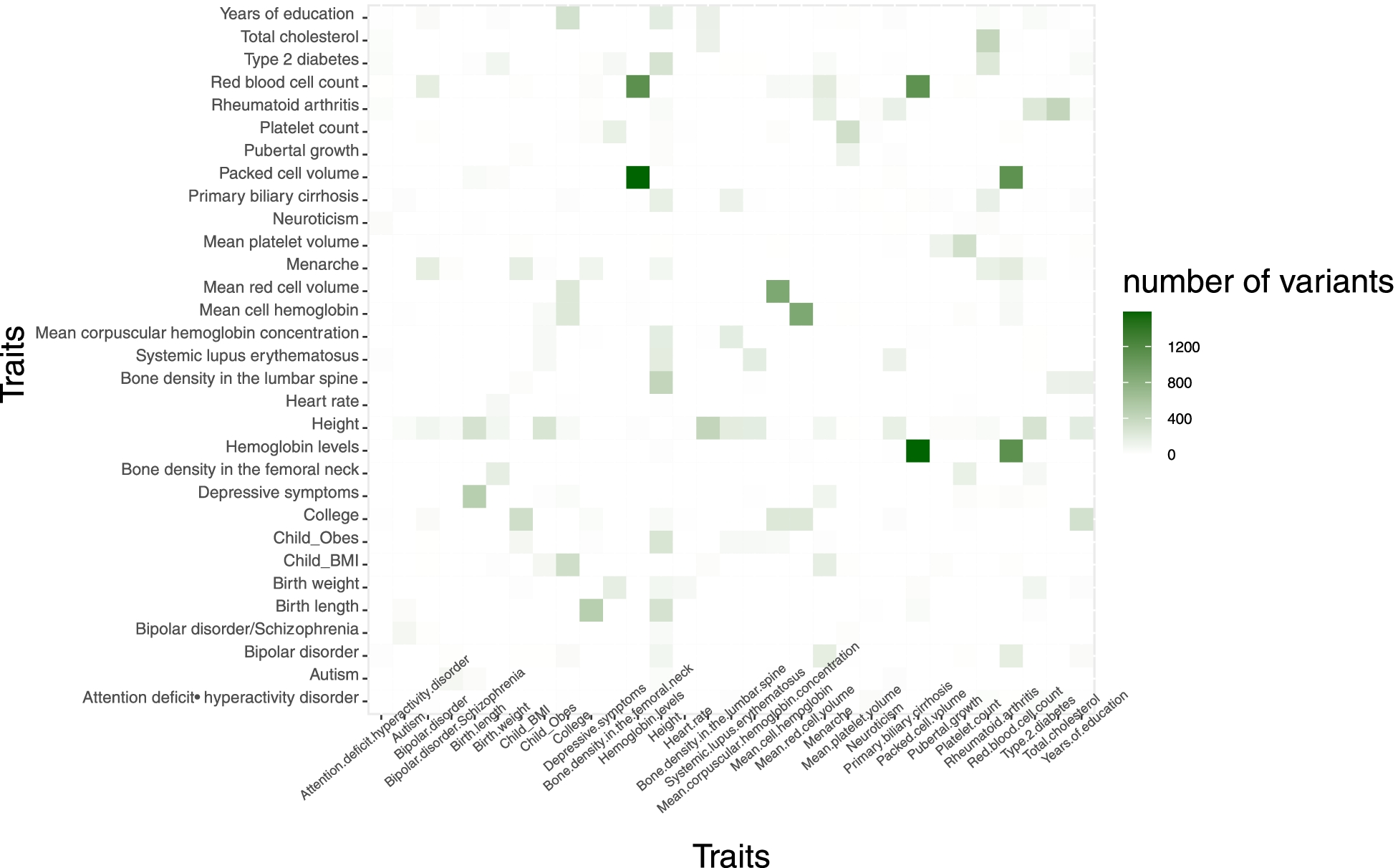
Materials and Methods We employed MIGWAS, ANNOVAR, MEME, and DAVID software packages to annotate the variants obtained from GWAS for 31 traits and elucidate the association between their harboring genes and their related traits. We identified variants that occurred in the motif regions that may be functionally important in affecting miRNA binding. We also conducted pathway analysis and functional annotation on miRNA targeted genes harboring 3’UTR variants for a trait with the highest percentage of 3’UTR variants occurring.
Results The Child Obesity trait has the highest percentage of 3’UTR variants (75%). Of the 16 genes related to the Child Obesity trait, 5 genes (ETV7, GMEB1, NFIX, ZNF566, ZBTB40) had a significant association with the term DNA-Binding (p < 0.05). EQTL analysis revealed 2 relevant tissues and 10 targeted genes associated with the Child Obesity trait.
Conclusion Variants located in 3’UTR can alter the binding affinity of miRNA and impact gene regulation, thus warranting further annotation and analysis. We have developed a bioinformatics bash pipeline to automatically annotate variants, determine the number of variants in different categories for each given trait, and check common variants across different traits. This is a valuable tool to annotate a large number of GWAS result files.