Abstract
Overview
Identifying genetic variants that are associated with variation in DNA methylation, an analysis commonly referred to as methylation quantitative trait locus (meQTL) mapping, is an important first step towards understanding the genetic architecture underlying epigenetic variation. Most existing meQTL mapping studies have focused on individuals of European ancestry and are underrepresented in other populations, with a particular absence of large studies in populations with African ancestry
Results
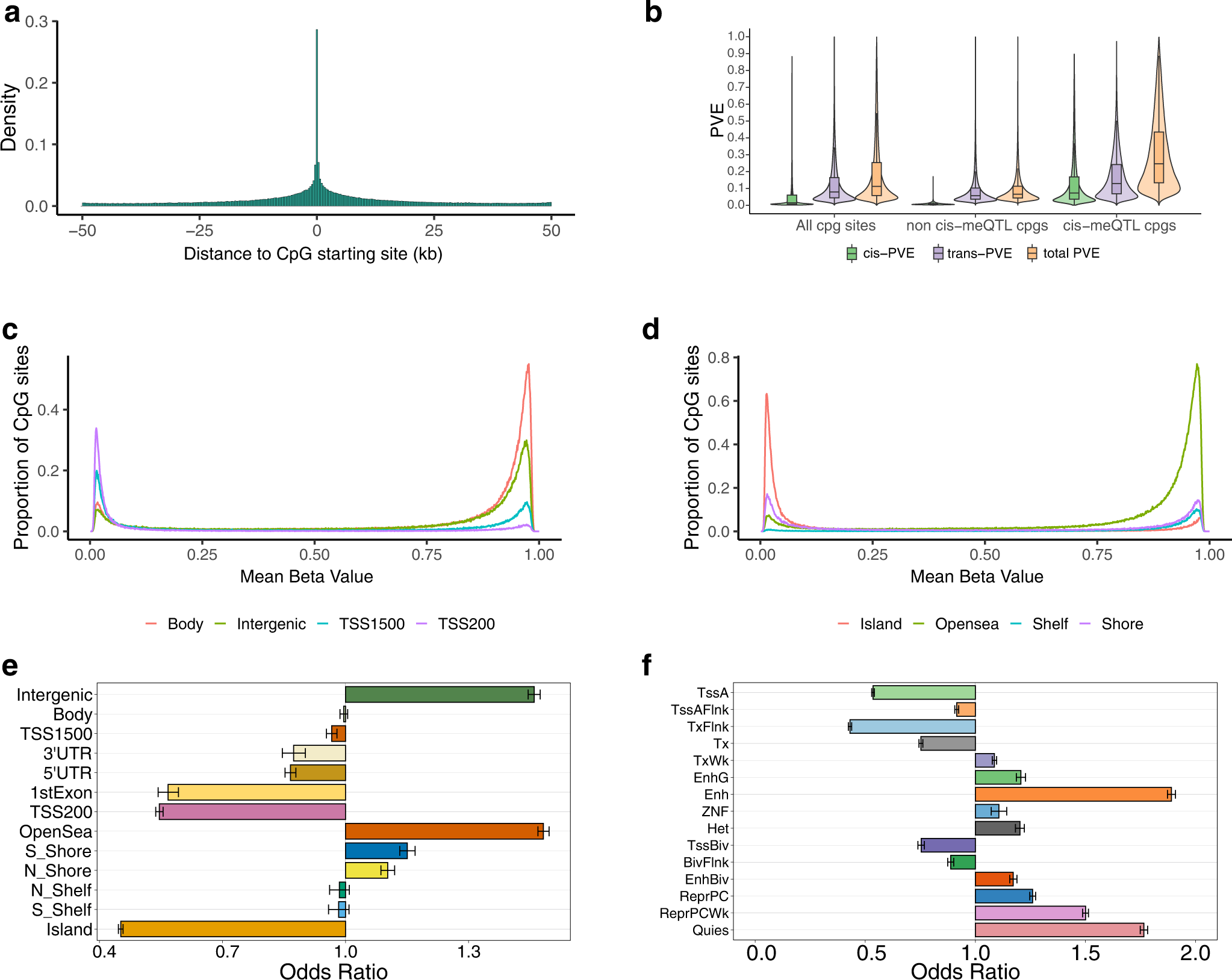
We identify a total of 4,565,687 cis-acting meQTLs in 320,965 meCpGs. We find that 45% of meCpGs harbor multiple independent meQTLs, suggesting potential polygenic genetic architecture underlying methylation variation. A large percentage of the cis-meQTLs also colocalize with cis-expression QTLs (eQTLs) in the same population. Importantly, the identified cis-meQTLs explain a substantial proportion (median = 24.6%) of methylation variation. In addition, the cis-meQTL associated CpG sites mediate a substantial proportion (median = 24.9%) of SNP effects underlying gene expression.
Conclusion
Overall, our results represent an important step toward revealing the co-regulation of methylation and gene expression, facilitating the functional interpretation of epigenetic and gene regulation underlying common diseases in African Americans.
DOI
https://www.nature.com/articles/s41467-023-37961-4
Citation
Shang, Lulu, Wei Zhao, Yi Zhe Wang, Zheng Li, Jerome J. Choi, Minjung Kho, Thomas H. Mosley, Sharon LR Kardia, Jennifer A. Smith, and Xiang Zhou. “meQTL mapping in the GENOA study reveals genetic determinants of DNA methylation in African Americans.” Nature Communications 14, no. 1 (2023): 2711.